🔑 Key Learning
-
Wilms tumour is the most common renal tumour in childhood, presenting as a painless abdominal mass, haematuria and flank pain in children < 4 years.
-
Neuroblastoma arises from neural crest cells of the sympathetic nervous system, often secretes catecholamines and presents variably depending on site.
-
Retinoblastoma presents in children < 3 years, often with leukocoria and strabismus, and is linked to RB1 tumour suppressor gene mutations.
-
All three conditions require urgent specialist referral for further investigation and management.
🥬 Wilms Tumour (Nephroblastoma)
Pathophysiology
-
The most common renal tumour in childhood (5% of all childhood malignancies).
Clinical Features
-
Age < 4 years in 75% of cases.
-
Painless, rapidly growing abdominal mass.
-
Haematuria.
-
Hypertension.
-
Flank pain or fever.
Red Flags & Referrals
-
Palpable abdominal mass or organomegaly OR unexplained visible haematuria – very urgent referral (within 48 hours).
Investigations
-
Abdominal ultrasound (initial test).
-
Followed by CT or MRI for staging.
Management
-
Neoadjuvant chemotherapy followed by surgical resection.
-
Further chemotherapy ± radiotherapy based on staging.
🧠 Neuroblastoma
Pathophysiology
-
Malignant tumour of sympathetic nervous system.
-
Derived from neural crest cells.
-
Common primary sites: adrenal medulla, sympathetic ganglia (abdomen, thorax, pelvis, neck).
Clinical Features
-
Usually diagnosed at around 2 years of age.
-
Abdominal mass: pain, distension.
-
Thoracic mass: respiratory symptoms, dysphagia.
-
Cervical tumour: Horner’s syndrome.
-
Metastases to bone marrow: fatigue, pallor, bruising.
-
Systemic symptoms due to catecholamine secretion: flushing, sweating, tachycardia, hypertension.
Red Flags & Referrals
-
Palpable abdominal mass or organomegaly – very urgent referral (within 48 hours).
Investigations
-
Urine catecholamines: elevated VMA/HVA levels.
-
Imaging: CT/MRI.
-
Biopsy to confirm diagnosis.
Management
-
Risk-adapted multimodal therapy: surgery, chemotherapy, radiotherapy, immunotherapy, stem cell transplant (if high-risk).
👁️ Retinoblastoma
Pathophysiology
-
Malignancy of retinal cells.
-
Caused by mutations in the RB1 tumour suppressor gene on chromosome 13.
Genetic Subtypes
-
Germline mutation: bilateral tumours, high risk of additional malignancy (e.g. osteosarcoma).
-
Sporadic mutation: unilateral tumour, no increased risk of other cancers.
Clinical Features
-
Presents < 3 years (often < 1 year if bilateral).
-
Leukocoria – white pupillary reflex.
-
Strabismus (squint).
-
Decreased visual acuity.
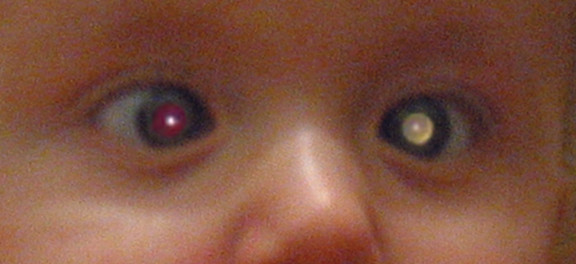 Retinoblastoma. Leukocoria.
Retinoblastoma. Leukocoria.
Red Flags & Referrals
-
Absent red reflex – urgent referral (suspected cancer pathway).
Investigations
- MRI orbit/brain (for extent).
Management
-
Chemotherapy (systemic or intra-arterial).
-
Local therapies (cryotherapy, laser photocoagulation).
-
Enucleation if extensive disease.
📝 Exam Clues & Clinchers
-
Toddler with painless abdominal mass, haematuria and HTN → Wilms tumour → US abdomen, very urgent referral.
-
Flushing, abdominal mass, bone marrow failure signs in child < 5 → Neuroblastoma → urine VMA/HVA.
-
White pupil in infant photo → Retinoblastoma → absent red reflex → urgent ophthalmology referral.
